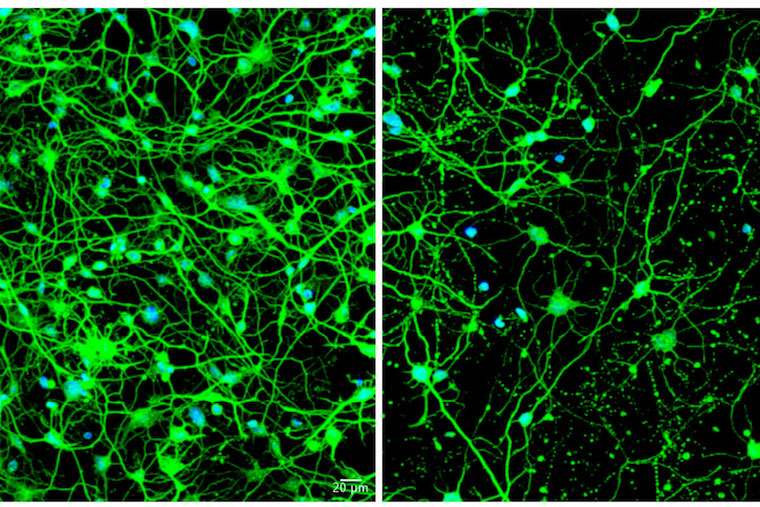
Huntington’s disease, a fatal, inherited neurodegenerative condition, is caused by a genetic error present at birth, though its symptoms often don’t begin until middle adulthood. Scientists at Washington University School of Medicine in St. Louis have been trying to understand how the aging process triggers the onset of symptoms, with the expectation that such knowledge could point to treatments that delay or prevent neurodegeneration.
To that end, a new study from Washington University indicates that as patients age, the disease gradually impairs an important cellular housekeeping process called autophagy, which is responsible for eliminating waste from cells. This housekeeping is significant in Huntington’s because a buildup of waste in a specific kind of neuron leads to such cells’ untimely deaths.
The researchers also showed that enhancing the autophagy pathway in such neurons that were created from skin cells of Huntington’s patients protects those cells from dying.
“Our study reveals how aging triggers a loss of the crucial process of autophagy — and hints at how we might try to restore this important function, with the aim of delaying or even preventing Huntington’s disease,” said senior author Andrew S. Yoo, PhD, a Washington University professor of developmental biology.
The study, published Oct. 27 in the journal Nature Neuroscience, also may offer clues to understanding cognitive decline in aging generally.
Huntington’s disease destroys a specific type of brain cell called medium spiny neurons, the loss of which causes involuntary muscle movements, impaired mental health and cognitive decline. Patients typically live about 20 years after signs of the disease first appear.
For this study, the researchers reprogrammed patients’ skin cells into medium spiny neurons using a technique they developed that allows adult skin cells to be transformed directly into various types of brain cells, depending on the specific recipe of signaling molecules to which the skin cells are exposed. More common techniques involve use of stem cells — but stem cells reset the cells’ biological clocks to an early developmental state, which is not useful when studying diseases that only become symptomatic in adulthood.
“We collected skin cell samples from different patients at a range of ages and modeled the disease before and after symptoms developed, which allowed us to identify the differences between younger and older patients with Huntington’s disease,” Yoo said. “We knew there must be some change that takes place as patients age. They all have a genetic mutation in the Huntingtin gene. We wanted to find the difference between young patients who have no symptoms and older patients who actively show signs of the disease.”
Yoo and his colleagues, including co-first authors Youngmi Oh, PhD, and Seongwon Lee, PhD, both staff scientists in Yoo’s lab, found that medium spiny neurons reprogrammed from skin cells of older patients with symptomatic Huntington’s produced very high levels of a microRNA molecule called miR-29b-3p. These high levels were not seen in reprogrammed neurons of younger Huntington’s patients or in reprogrammed neurons from healthy individuals of any age. The investigators showed that the microRNA set off a chain of events that included impairing autophagy in these cells. When the skin cells completed the conversion into neurons, they began producing the problematic microRNA, autophagy slowed down, and the cells began dying.
The researchers went on to show that reducing levels of this microRNA allowed autophagy to continue and protected the neurons from dying. In addition, they found that enhancing autophagy with a chemical compound called G2 protected the diseased neurons from death. As the researchers increased the dose of G2, the protection from cell death improved as well.
G2 is derived from a series of analogs that were discovered in the labs of co-authors David Perlmutter, MD, executive vice chancellor for medical affairs, the George and Carol Bauer Dean of the School of Medicine, and the Spencer T. and Ann W. Olin Distinguished Professor; Gary Silverman, MD, PhD, the Harriet B. Spoehrer Professor and head of the Department of Pediatrics; and Stephen C. Pak, PhD, a professor of pediatrics in the Division of Newborn Medicine. G2 was identified via high throughput screening for autophagy enhancer drugs that could correct the cellular accumulation of variant alpha-1-antitrypsin Z that causes liver disease in alph-1-antitrypsin deficiency (ATD). The G2 compounds could therefore represent attractive candidates for preventing neurodegeneration in Huntington’s disease, liver disease in alpha-1-antitrypsin deficiency and perhaps other diseases in which aberrant accumulation of misfolded proteins is toxic to cells.
The study also uncovered what may be a tantalizing clue for understanding cognitive decline in normal aging. When comparing the symptomatic neurons to pre-symptomatic neurons and to healthy neurons from both young and older adults, the researchers found that the neurons of healthy older adults produced slightly elevated levels of the harmful microRNA, but in far smaller amounts than the neurons of symptomatic Huntington’s disease patients. The study suggests that even in normal, healthy aging, medium spiny neurons gradually produce low levels of this microRNA, which may interfere with autophagy’s healthy cellular housekeeping.
“By modeling different stages of the disease across the life span, we can identify how aging plays a role in disease onset,” Yoo said. “With that information, we can begin to look for ways to delay that onset. Our study also suggests that the triggering molecule for the onset of Huntington’s disease may play some role in age-associated decline in neuronal function generally. Understanding the component of aging that sets off neurodegeneration may help in developing new strategies for treatment and prevention of Huntington’s disease and other neurodegenerative conditions that develop at older ages.”
Yoo and his team also are working with other collaborators using their cellular reprogramming technique to investigate forms of Alzheimer’s disease, tauopathy, and other neurodegenerative conditions.
This work was supported by the National Institutes of Health (NIH), grant numbers RF1AG056296 and R01NS107488; the Cellular and Molecular Biology Training Program, grant number T32 GM007067; the Cure Alzheimer’s Fund; the CHDI Fund; a Hereditary Disease Foundation Grant; the Farrell Foundation Fund; and a Mallinckrodt Scholar Award.
Oh Y, Lee S, Kim WK, Chen S, Church VA, Cates K, Li T, Zhang B, Dolle RE, Dahiya S, Pak SC, Silverman GA, Perlmutter DH, Yoo AS. Age-related Huntington’s disease progression modeled in directly reprogrammed patient-derived striatal neurons highlights impaired autophagy. Nature Neuroscience. Oct. 27, 2022.
About Washington University School of Medicine
WashU Medicine is a global leader in academic medicine, including biomedical research, patient care and educational programs with 2,700 faculty. Its National Institutes of Health (NIH) research funding portfolio is the fourth largest among U.S. medical schools, has grown 54% in the last five years, and, together with institutional investment, WashU Medicine commits well over $1 billion annually to basic and clinical research innovation and training. Its faculty practice is consistently within the top five in the country, with more than 1,790 faculty physicians practicing at over 60 locations and who are also the medical staffs of Barnes-Jewish and St. Louis Children’s hospitals of BJC HealthCare. WashU Medicine has a storied history in MD/PhD training, recently dedicated $100 million to scholarships and curriculum renewal for its medical students, and is home to top-notch training programs in every medical subspecialty as well as physical therapy, occupational therapy, and audiology and communications sciences.