Anyone who follows science has read enthusiastic stories about medical breakthroughs that include the standard disclaimer that the results were obtained in mice and might not carry over to humans.
Much later, there might be reports that a drug has been abandoned because clinical trials turned up unforeseen side effects or responses in humans. Given the delay, most readers probably don’t connect the initial success and the eventual failure.
But Igor Efimov, PhD, a biomedical engineer at Washington University in St. Louis who studies the biophysical and physiological mechanisms that underlie heart rhythm disorders, is acutely aware of the failure of once-promising drugs to pass clinical trials.
“The problem is the difference in gene expression between the mouse and the human is very very large,” Efimov says.
Mice are the most popular animal model in cardiovascular research in part because it is easy and cheap to create a transgenic mouse, and these mice allow research questions to be asked and answered precisely and quickly.
Two recent research studies have found differences between the distribution of potassium-ion-channel variants in the mouse heart and in the human heart. In the mouse, the ion channels in the atria are different from those in the ventricles. In humans there is no such chamber specificity. The difference is crucially important for the development of safe and effective cardiovascular drugs.
To avoid the “mouse trap,” Efimov has established connections with local institutions that supply his lab with human hearts. The hearts are either diseased ones removed from patients undergoing heart transplants or “non-failing” hearts that have been donated for research but are considered unsuitable for transplantation.
An article in the August issue of the Journal of Molecular and Cellular Cardiology beautifully demonstrates the importance of working with human hearts. It reports on studies in human hearts of two drugs that had already been studied in the mouse heart (work also published in the Journal of Molecular and Cellular Cardiology). The results show that a drug target that looked promising in the mouse model would not work in humans.
The research is a collaboration between Efimov, the Lucy & Stanley Lopata Distinguished Professor of Biomedical Engineering, Cell Biology and Physiology, and Radiology, and Colin G. Nichols, PhD, the Carl Cori Professor of Cell Biology and Physiology and co-director of the Center for the Investigation of Membrane Excitability Disorders (CIMED) at the School of Medicine.
“We were able to demonstrate this without the expense of clinical trials and without putting patients at risk,” Efimov says.
The KATP ion channel
The heart is an electromechanical pump, and normal contraction of the heart muscle depends on normal electrical activity. Electrical activity in the heart ultimately reflects the coordinated opening and closing of ion channels in the surface membranes of heart muscle cells.
Ion channels are proteins that control the passage of electrically charged atoms, called ions, into and out of cells. The currents that flow through the heart with each beat depend on the orchestrated opening and closing of channels specific for sodium, potassium and calcium ions. Genetic mutations that affect ion-channel function are thought to underlie many forms of heart disease.
Efimov and Nichols have been studying an ion channel known as the KATP channel. This is a potassium channel that is sensitive to the presence of ATP, the body’s energy storage molecule.
The KATP channel is both a tempting and dangerous target for drug therapy. When there is a blockage in the coronary artery, the main vessel that carries blood to the heart muscle, this channel immediately opens. By opening, says Efimov, the channel seems to protect the heart from oxygen deprivation.
On the other hand, the activation of the channel can kill the patient by dramatically shortening the action potentials, or electrical impulses that trigger heart-muscle contraction. This shortening causes the onset of arrhythmia, or the loss of normal heart rhythm.
The mouse heart
The KATP channel can have either of two regulatory subunits that are sensitive to the presence of ATP. These are called SUR1 and SUR2 (for sulfonylurea receptor types 1 and 2).
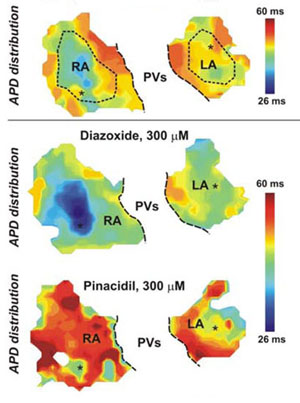
The mouse heart’s response to two cardiovascular drugs. By bathing heart tissue in voltage-sensitive dyes that fluoresce with an intensity proportional to voltage scientists are able to map the duration of nerve impulses in the heart. These atrial maps correspond to untreated hearts (top), hearts perfused with the drug diazoxide (middle) and those treated with the drug pinacidil (bottom). The effect of the drugs was chamber specific: diazoxide decreased action-potential duration in the atria, but pinacidil had little effect on them.
In an article published in the January 2010 issue of the Journal of Molecular and Cellular Cardiology, Nichols and Efimov reported that, in the mouse, the SUR1 gene is expressed only in the atria and not in the ventricles. SUR2, on the other hand is expressed only in the ventricles and not in the atria.
“This is really quite remarkable,” Efimov says, “because there are very good drugs that are specific to SUR1 or to SUR2. This means if the mouse data translates to humans, it would be possible to design a drug that would work only on the atria without affecting the ventricles.”
“The lack of specificity is a huge problem with other drugs,” he says. “If you want to treat atrial fibrillation, you need a drug that works only on the atria and does not affect the ventricles. If you treat with a drug that has some targets in the ventricles, you’ll treat the atrial fibrillation, but you’ll kill by ventricular fibrillation.”
Fibrillation is the chaotic or unsynchronized contraction of heart muscle. Ventricular fibrillation causes sudden cardiac death because the heart no longer pumps effectively and blood does not reach the brain. Atrial fibrillation is less lethal but puts patients at increased risk from stroke and sudden cardiac death.
The human heart
After the mouse study, Efimov and Nichols wanted to know if the results would hold in the human heart.
To find out, the scientists repeated the mouse study with human hearts. They excised a piece of the heart that included both atrial and ventricular tissue, perfused it with a solution that would keep the tissue functioning and treated it with two drugs. One is specific to the SUR1 variant of the potassium-ion channel, and the other is specific to SUR2 variant.
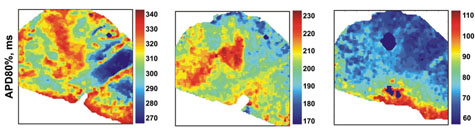
The human heart’s response to the same cardiovascular drugs. These atrial maps of failing hearts show the response without drugs(left), to the the drug diazoxide (middle) and to pinacidil (right). Both drugs significantly reduced action-potential duration in the atria. (Note that each map has a different color scale.) What holds for the mouse heart, does not hold for the human heart.
In order to record the drugs’ effects, they bathed the heart tissue with voltage-sensitive dyes. These bind to the membrane of cardiac cells and, when illuminated with an arc lamp, fluoresce with an intensity directly proportional to the transmembrane voltage.
Action potential duration (the physiologically important parameter) can be calculated from the change in these intensities over time.
The result? “In human hearts,” Efimov says, “it doesn’t work. The SUR1 drug doesn’t work in the atria at all, but it does affect the ventricles; it is the opposite of what happens in the mouse. The SUR2 drug affected both the atria and the ventricles, and shortened the action potentials in the ventricles so much that it would cause fatal arrhythmias in people.”
A better paradigm
Efimov sees the results as indicative of a larger problem with cardiovascular research, one that has blocked the development of effective therapies for many years.
The current approach to studying arrhythmia and many other diseases goes back to a three-step protocol worked out by the German scientist Rudolf Virchow, Efimov says. The first step is to identify the clinical signs and symptoms of the disease; the second is to recreate those symptoms and identify a therapy in an animal model; and the third evaluate the safety and efficacy of the therapy in clinical trials.
“The problem is that at least in the cardiac arrhythmia field, this paradigm has had very few successes,” Efimov says. “It has resulted in the discovery of almost no successful drugs. Clinical trial after clinical trial has ended in failure.”
Mice are the most popular animal model in physiology, but the mouse is not a very good model for cardiac physiology. “A mouse’s heart beats about 600 times per minute, so you can imagine it is a little different from humans, whose hearts beat on average 72 times per minute,” Efimov says.
“You can mutate in mice the gene thought to cause heart failure in humans and you don’t get the same disease, because the mouse is so different,” Efimov says.
“So, unfortunately, even with the help of transgenic mice, very few results made it from the animal model to the clinic.”
The answer, Efimov says, is to insert an additional step in the three-step research protocol. After a therapy has been tested in an animal model, it should be tested in human hearts before moving to clinical trials.
“Since we’ve begun to work with human hearts,” he says, “we’re finally starting to catch up with animal physiology.”